
| |
| Volume 10, Number 16 | November 18, 2020 |
The two-phase theory of human aging was applied to modern actuarial data in the last issue.[1] The theory was found capable of explaining the detailed pattern of these data. This appears to be the first time any theory of aging has been able to explain these data, thus corroborating the two-phase theory.
The two-phase theory says that modern human aging is a syndrome of three diseases:[2]
Aging 0: congenital vitamin MePA (methyl-phosphonic acid) deficiency disease,
Aging 1: congenital vitamin MePiA (methyl-phosphinic acid) deficiency disease, and
Aging 2: a mitochondrial genetic disease induced by Aging 1.
When the two-phase theory of human aging was introduced, and the discovery of Aging 2 was announced, the need for a biological timed switch was highlighted. A biological timed switch is needed to explain the form of the ancient biblical life span data.[4] Also highlighted was the need for a biological Poisson process to provide the needed biological timer component of the timed switch. In the six months which have intervened since the introduction of the two-phase theory, I have found it surprisingly difficult to come up with a suitable biological Poisson process.
A suitable Poisson process, in the present context, will involve sequential events whose exact timing from one event to the next is randomly distributed around some average time.
Imagine unlocking three equally spaced doors. Suppose the doors are side by side: Door 1 next to Door 2 next to Door 3. Then the time it takes to unlock the three doors will depend on the order in which the doors are unlocked. The order 1–2–3 will be faster than 1–3–2, for example, because of the extra time it takes to walk from Door 1 to Door 3 and then back to Door 2. The time it takes to unlock each of the three doors in this case is not randomly distributed in time—it depends on the order in which the doors are unlocked—so the unlocking of the side-by-side doors is not a Poisson process.
Now suppose that the three doors are located behind one another, blocking passage down a hallway, for example. It is then necessary to unlock Door 1 before one can get to Door 2 to unlock it, which then allows access to Door 3 to unlock it. In this case, the time it takes to unlock each door will be randomly distributed around some average door-unlocking time, and the unlocking of these sequential doors will be a Poisson process.
The significance of this, in the present context, is that such a process allows one to design a mechanically-timed switch.
I have previously used the bursting of sequential water-filled balloons by pellets from an air rifle as an example of a mechanically timed switch.[5]
Imagine a sequential set of N balloons tied with strings in a row on a slanted rod. When the end balloon gets hit and bursts, the next balloon slides down and takes its place. When the last water balloon is burst, the rod tips up, flipping a switch to start some process. The average total time, T, for multiple attempts at bursting all of the balloons and flipping the switch will be N times the average number of shots it takes to burst a balloon divided by the rate at which pellets are fired. Because this is a Poisson process, the spread in total times (i.e., the standard deviation of total time measurements) will be √ T .
If there are 10 balloons and the rifleman takes 10 shots on average to hit one balloon and fires at 1 pellet per minute, then it is possible to predict that he will take 100±10 minutes start to finish to flip the switch.
The biblical life span data for pre-Flood males say that the age of onset of Aging 2 is controlled by such a mechanically-timed switch.
The age of onset of Aging 2 pre-Flood was 800 years. Today it is probably within two years of age twelve. Clearly, the timer is running faster today than it was in the ancient past—the bullets are flying faster than they were back then.
The two-phase theory says that this is due to loss of vitamin MePiA from modern human diets. Vitamin MePiA is an antioxidant. The theory says that this vitamin protects mitochondrial DNA (mtDNA) from damage due to free radicals.[6] The bullets correspond to free radicals, and the bursting of the balloons corresponds in some way to free radical damage of mtDNA. This much is known.
Also known is that the number of balloons is large. The full width at half maximum of the spread in age of death back before the Flood was 49 years. Today it is 28 years. This allows the number of balloons to be estimated. One finds a minimum of 2,200 balloons.[7]
What is not known—what the present issue is meant to reveal—is the exact biological process responsible for the timed switch in the case of Aging 2. Suitable processes must be of the Poisson type, and these are evidently not abundant. In fact, the example originally given—the shortening of telomeres—is the only candidate which I have been able to discover.
For supplying a Poisson sequence, the telomere candidate works nicely.
Recall that telomeres are non-coding bits of DNA which provide the ends of chromosomes with a protective cap. Recall also that shortening of telomeres happens each time the cell divides and the chromosomes need to be replicated. Thus, shortening of a given telomere is a sequential process, and, like the sequentially burst balloons, the ordering of events is physically constrained. The second shortening can happen only after the first shortening has happened, etc. Taking cell replication for the adult body to be randomly distributed around some average replication time, a Poisson sequence results for a given chromosome from the times between the shortening of its telomeres.
A further boost for this candidacy is that the fundamental, repeated, coded sequence for telomeres, TTAGGG, "is usually repeated about 3,000 times."[8] This implies a potential for about 3,000 balloons per telomere—which is immediately in the same ballpark as the minimum 2,200 needed by the Aging 2 timed switch. A large number of balloons is evidently not a problem for telomeres.
But there is a major problem with the telomere candidacy. As was noted in the original article,
Human mtDNA does not have telomeres. Human mtDNA is a circular molecule having no ends needing to be capped. Clearly, something other than telomere shortening is going on with mtDNA.[9]
This yields a conundrum. The theory says that Aging 2 is a mitochondrial genetic disease. To begin to explain Aging 2 biologically seems to require that telomeres exist on human mtDNA, but human mtDNA is a circular molecule and thus has no ends and no end caps—no telomeres.
The only way I have been able to find to solve this conundrum is via the bold postulate that human mtDNA must possess "in-line telomeres" (Figure 1). I call this postulate "bold" because I have never seen or heard of such a thing as "in-line telomeres," making them to be newly invented, purely hypothetical entities.
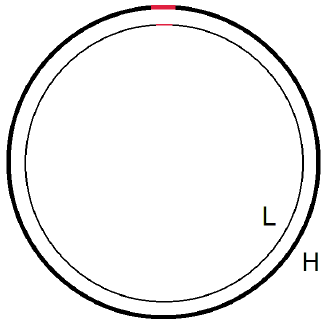 |
The first and most obvious question which this bold postulate raises is, "What are 'in-line telomeres' for—what purpose would they serve?"
Actually, this question needs to be flipped on its head. A much bigger and more obvious question is, "How could human mtDNA manage to make do without any telomeres?" Allow me to explain the problem.
Maintenance of the blueprints is necessarily of highest priority for any organism.
A house is constructed by working from a set of blueprints. The blueprints code the idea of the building. Change the blueprints and the idea changes and a different building gets constructed. The physical building is simply the tangible expression of the idea coded by the blueprints.
Living organisms are also constructed from blueprints. While house blueprints are made of paper and ink, biological organisms' blueprints are made of DNA.
The DNA codes the idea of the organism. The fish idea is quite different from the bird idea, for example, and the DNA code for the two is quite different.
In writing, we use words and sentences to code ideas. Here is an example of an idea coded in writing using a typewriter:
The box was red.We could use writing to code the idea for a house we wished to have built. We don't normally do so because the idea is much more easily and efficiently coded by means of a drawing—a blueprint.
Now here is the important point in the present context. Coded ideas are rather fragile things.
Watch what happens to the idea
The box was red.if I accidentally make a simple typing error while coding it:
The fox was red.Notice that the original idea has vanished and been replaced by a whole new idea.[10] A tiny coding error can be majorly destructive of a coded idea.
Most coding errors result in something unintelligible, like this:
The gox was red.In this case, the original idea has vanished and been replaced by gibberish.
For cells, coded gibberish represents erosion of the idea of the organism with concomitant loss of functionality. Cells are marvelously resourceful and able to withstand some loss of function, but the principal expectation of accumulating gibberish is eventual death of the cell.
For this reason, maintaining the fidelity of its DNA code is necessarily of highest priority for any organism.
Now, DNA risks loss of fidelity every time it is replicated. Replication is carried out by dedicated copy machinery within the cell. While this replication machinery is extraordinarily good—much better than any human typist—it is not perfect. For human mtDNA, the error rate is less than one mistake per million "letters" of the code copied.[11] This is a very low error rate, but when you consider that all humans start out as a single cell and wind up being comprised of about 37 trillion cells, which then get replaced every 7 to 10 years, DNA copy errors are clearly a problem which must somehow be addressed if the organism is to survive and prosper.
How might a self-replicating biological machine be designed to minimize the impact of unavoidable mtDNA copy errors?
If you ponder this question for a while, I think you will come to the conclusion that it is necessary, when making a new mtDNA copy, to use as a template the lowest generation number copy available.
Take the stem cell to be generation 0. The first mtDNA replication gives rise to a generation 1 copy. If the generation 1 copy is used as a template, the next new copy will be generation 2. If the generation 0 copy is used as a template again, one gets another generation 1 copy, distinct from the first generation 1 copy. Since each "letter"-copying event has a non-zero probability of producing a copy error, a generation 2 copy is likely to contain more copy errors than a generation 1 copy. A generation 2 copy will contain both the copy errors the generation 1 template had (which the copying machinery has copied to generation 2) plus any new copy errors made in the production of the generation 2 copy.
To suppress the spread of copy errors, best design calls for the use of the lowest generation template available when making a new copy.
Now, if you think on this a little longer, I think you will find that the only way to implement this best design strategy is to tag each and every mtDNA copy with a generation number, so the lowest generation number copy can be selected from available copies for use as the template for making the next new copy.
Telomeres provide precisely this sort of tagging function. They not only cap and protect the ends of nDNA, they also keep track of how many times a cell has replicated.
Now you can see the problem. How does human mtDNA manage to get along, as the organism progresses from 1 initial cell to over 37 trillion cells, without this telomere function of keeping track of the generation number so copy errors can be suppressed?
I suggest that the function of "in-line" telomeres is to keep track of the generation number of each mtDNA copy.
This generation-counting function for "in-line telomeres" allows this new, hypothetical entity to be christened with a new name. I reached out to a friend, Tom Godfrey (PhD linguistics), for help with this. He suggested the Greek word "psephos." (The leading p is silent.)
Another English word, psephology, contains a good candidate. The initial Greek root in this word refers to a pebble. The Greek verb for the action of counting has the same root (https://en.wikipedia.org/wiki/Psephos), evidently because pebbles were once used for voting or counting. You may already know that calculus means small pebble in Latin, but I think you want to stick with Greek.[12]When I then asked about a "mere" ending, to place this entity in the same class as the telomere, Tom replied, "The Greek roots in telomere mean end and part, respectively, so psephomere works out to pebble/counter part."
"Psephomere" seems well suited to this mtDNA mechanical generation counter, based, as it is, on physical tokens (i.e., short bits of nucleotides). I will use it in place of "in-line telomere" from now on.
A second obvious question is, "Why is no psephomere evident in the published map of human mtDNA?"[13]
The answer to this question requires a second bold postulate. My second bold postulate is that the published map of human mtDNA is for mtDNA which has shortened its psephomere to zero.
I call this postulate "bold" because it seems so unlikely at the outset. But, here again, this flips on its head. As soon as this postulate is advanced, a complete theory of Aging 2 suddenly emerges, and this theory immediately shows that mtDNA which has shortened its psephomere to zero is normal (I do not mean to imply healthy) today, so that a modern map of mtDNA is most likely to show the zeroed-psephomere molecule. Said simply, the zeroed-psephomere molecule is Aging 2, and because Aging 2 disease is presently normal to the global population for all individuals past roughly age 10 years, randomly chosen mtDNA is most likely to be of the zeroed-psephomere type.
The newly emerged theory of Aging 2 looks as follows.
Psephomeres keep count of the generation number of every mtDNA copy, enabling suppression of mtDNA copy errors. The count is kept, as in the case of telomeres, by successively shortening the psephomere of each new mtDNA copy. Because generation 0 psephomeres are of finite length, excessive copying of mtDNA (due, for example, to a high free radical damage incidence necessitating frequent replacement of mtDNA copies) will result in eventual loss of psephomeres from the mtDNA population. These are Aging 2 diseased mtDNA copies. Loss of psephomeres breaks generation number counting, which breaks copy error suppression, which allows mtDNA copy errors (i.e., mtDNA genetic point mutations) to multiply, which results in exponentially progressing clinical Aging 2 disease over ensuing decades. That is, the expression of accumulated mtDNA copy errors results in increasing mitochondrial dysfunction, which results in increasing cellular dysfunction, which results in increasing whole organ and whole organism dysfunction, culminating ultimately in death of the individual.
In this theory, the shortening of the psephomere to zero is the long-sought mechanical biological timer, and the final decrement to zero of the psephomere is the associated switch which initiates Aging 2. (This theory displaces the mtDNA scrapping hypothesis previously advanced.)[14] The time required to shorten the psephomere to zero will depend on the rate of replacement of mtDNA, which will depend on the rate of damage of mtDNA due to free radicals, which will depend on cellular concentrations of vitamin MePiA. The depletion of the psephomere to zero breaks the generation counting function, which switches the mtDNA copy from healthy to Aging 2 diseased.
One of the important discoveries about the nature of human aging today resulting from the application of the two-phase theory of human aging to modern U.S. actuarial life table data in the last issue was that the exponential progression of Aging 2 disease slows down and begins to level off somewhat after 90 years of age. This is the newly discovered phenominon of Aging 2 saturation.
The theory of Aging 2 presented above harmonizes with the explanation of Aging 2 saturation ventured in the last issue, recommending its inclusion in the theory.
The primary role of mitochondria is energy production for the cell. Cells can produce energy not only via their mitochondria but also via glycolysis. While the role of glycolysis varies by cell type, in general, mitochondria carry most of the load of energy production for the cell, with the contribution from glycolysis being minor. But the existence of these two different energy sources means that cellular energy will not drop to zero as, due to exponentially increasing mitochondrial dysfunction, mitochondrial energy production drops to zero. Rather, cellular energy will drop no lower than the amount provided by glycolysis. Thus, the rate of increase of cellular dysfunction and death due to energy starvation, leading ultimately to whole organism dysfunction and death, will approach a constant saturation level even though mitochondrial dysfunction continues to progress exponentially.[15]
Mitochondria supply energy to cells by a process called oxidative phosphorylation (OXPHOS). Not surprisingly, the mtDNA genome is intimately involved with OXPHOS.
Mitochondrial DNA contains 37 genes, all of which are essential for normal mitochondrial function. Thirteen of these genes provide instructions for making enzymes involved in oxidative phosphorylation.[16]The implication of this intimate involvement is that the principal pathology stemming from onset of Aging 2 will be energy starvation of cells. Accumulating mtDNA copy errors will principally damage OXPHOS, resulting in diminished mitochondrial energy production, resulting ultimately in energy starvation.
The most obvious test, to check the veracity of this newly derived theory of Aging 2, is to look in young cells for healthy mtDNA. The younger the cells, the longer the psephomeres are expected to be.
Because this test requires specialized equipment and techniques, a result from it seems likely to be many weeks or months away.
Another test, which may be carried out immediately, is to look for vestiges of psephomeres on the map and associated replication algorithm of diseased mtDNA.
Imagine living on a remote island in the early 1920s. The island's population has heard about the wonders of the Model T Ford. Though never having seen one, they pool their resources and buy one. It arrives some months later, after a long sea voyage. Having never seen a Model T, the islanders fail to realize that the exposure to salt spray on the long sea voyage rusted the bolts holding the headlamps, causing the lamps to fall off and be lost at sea. The Model T is driven around the island for some weeks, but only during the day, since it is too dark to see where the car is going at night. Eventually, one islander gets the idea that headlights seem necessary for the car to be fully useful. He wonders why Henry Ford would fail to include them in the design of the car. He postulates that the car must originally have had headlights. How can he check his theory? He can look for headlamp vestiges on the car.
Going over the car carefully, he soon finds symmetrically placed sets of holes on the front of the car, one set on the left and one on the right. These holes seem to have no purpose. He surmises that they may have been put there to hold headlamps. Then he discovers broken-off wires dangling behind the holes. Tracing them, he finds that they run back to an on/off electrical switch on the dashboard. The islanders had wondered what the switch was for. Switching it on and off hadn't seemed to do anything. Now he has an explanation for the switch for the first time.
He takes the holes, the wires, and the switch—all essential components of an automotive headlamp system—to be vestiges of headlamps, and deems his theory, while not proven, to be confirmed beyond reasonable doubt.
Imagine a single mitochondrion containing several hundred mtDNA copies. Imagine that one copy gets damaged and needs to be replaced.
The mitochondrion is a highly complex biological machine, much more complex than an automobile, for example. It cannot think, but it can sense its own state and choose between various options depending on what it has detected. This ability to choose options depending on conditions should not seem shocking. The thermostat on your home furnace does the same thing all the time, and it is a very simple machine. It senses the air temperature in your home. If the air is too cold, it flips a switch calling for more heat. When the air gets warm enough, it flips the switch back, shutting off the heat. Think of the mitochondrion as a very complex machine equipped with many sensors and many switches controlling many processes.
The mitochondrion senses the irreparably damaged mtDNA copy and signals for it to be replaced. How the damaged copy is scrapped is not of interest in the present context. What is of interest is the replacement process. This process is, by hypothesis, designed to use the lowest generation copy in the mitochondrion as the template. How is the lowest generation copy selected, and how is it then replicated to yield a new copy with an appropriately shortened psephomere?
Before beginning to answer these questions, it is necessary to review what is known about human mtDNA replication. Only those details deemed to be important in the present context have been selected for review.
Each mtDNA copy is made up of two, distinguishable strands: the L strand and the H strand (Figure 2). These two strands may be thought of as parallel railway tracks. (The fact that, in real life, they spiral around each other as they go may be ignored for the present purpose.) The track goes in a circle. Think of the L strand as the inside track and the H strand as the outside track of this circle.
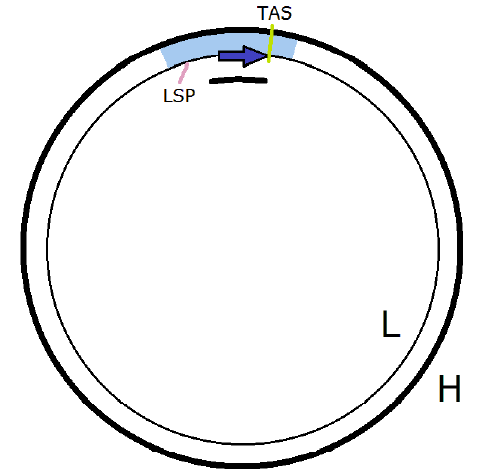 |
An important thing to get right at the outset is that it is not the case that a template mtDNA is simply copied during replication to yield the original template plus a new mtDNA copy. Rather, two new mtDNA copies are made, and the two-stranded template gets destroyed (used for parts) in making the copies. One of the template strands goes to Copy 1 and the other strand goes to Copy 2.
I will adopt the convention that the template L strand goes to Copy 1 and the template H strand goes to Copy 2. This is the order in which the two new copies are made—first Copy 1 and then Copy 2.
The mtDNA non-coding region (NCR) is of particular interest in the present context. The NCR takes up about 7% of the length of the track in the map of diseased mtDNA.
For purposes of copying mtDNA, mitochondria possess multiple copies of a special apparatus, called POLγ, which may be thought of as a monorail engine which rides one strand of a template mtDNA to make a new complementary strand, synthesizing one small segment of the complementary strand at a time. Synthesizing new strands of track is what POLγ is mostly, but not exclusively, about. It can also cut out segments of track, for example.
A side entrance to the NCR (called LSP, for light strand promoter) is situated toward one end of the NCR. It employs special apparatus to help POLγ get onto the track. This gets POLγ mounted on the L strand, facing down the track in the clockwise direction. POLγ is then set to begin scanning the L strand and synthesizing a new complementary H strand. When the new H strand is finished, these two strands will then become Copy 1.
POLγ is observed to halt at TAS (termination-associated sequence) 95% of the time rather than going full circle to complete Copy 1. TAS appears to be a gate which is regulated by the mitochondrion, keeping POLγ parked or letting it continue down the track synthesizing the new H strand. The reason for this gated behavior is uncertain.
Parking POLγ creates a short three-strand section of track called the "D-loop." The function or purpose of the D-loop is unknown.
When the TAS gate is opened, POLγ continues slowly around the track clockwise, scanning the template L strand to make the new complementary Copy 1 H strand as it goes (Figure 3). When it has completed about five eighths of the circular track, a mechanism automatically triggers for another POLγ to mount the template H strand to begin to copy it, producing its new Copy 2 L strand. The distance of five eighths—greater than one half of the circle—allows Copy 1 to be completed and the two template strands to be completely separated from each other so that POLγ 2 can complete its synthesis of Copy 2 without ever encountering—colliding with—the POLγ 1 synthesis.
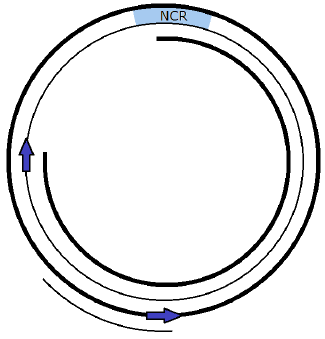 |
The synthesis of Copy 2 proceeds in a counterclockwise direction, back toward the NCR. Meanwhile, POLγ 1 completes its journey, encountering the starting end of the new complementary Copy 1 H strand it has been synthesizing. It is time to join the ends together. At this point, a curious behavior is observed with diseased mtDNA. POLγ is observed repeatedly to make and then delete the joint. This behavior is currently called "idling." From a design perspective, this idling behavior makes no sense. It seems inefficient at best and pointless at worst.
I suggest that idling may have been misnamed and misunderstood. I suggest that it may rather be "stuttering." It appears that it may be a vestige of the psephomere shortening step.
In a healthy mtDNA, once synthesis of the Copy 1 H strand has been completed, the new H-strand psephomere needs to be shortened to increment the generation counter. I suggest that on Aging 2 diseased mtDNA, POLγ 1 is unable to complete the psephomere-shortening step because the psephomere has already been shortened to zero. It tries—it deletes—but this triggers an error condition. So it rebuilds the deleted segment. Then it tries again, only to trigger the error condition again.
POLγ 1 sits there stuttering in this way because it is trying to deal with a worn out part. The psephomere has been used up. Eventually, it completes its preprogrammed number of delete cycles, which were intended to shorten the psephomere a fixed amount, and moves on.
This places the psephomere for healthy mtDNA at the very beginning of Copy 1, at the point where POLγ 1 first begins synthesizing the new H strand (Figure 4). This point is normally called OH, for the origin of H-strand replication. This location makes sense. It is in the noncoding region (the NCR), and like telomeres, psephomeres are noncoding.
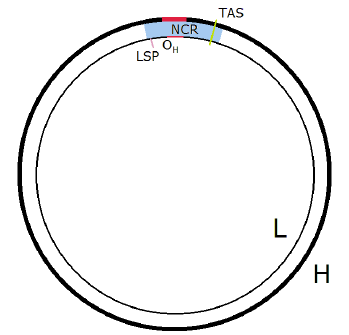 |
This also explains the need for a loop in this region. The shortening of the Copy 1 psephomere H strand will cause it to be shorter than the Copy 1 psephomere template L strand. To properly match the complementary H and L strands on either side of the shortened region requires that there be a loop to accomodate the extra L strand length.
The fact that Copy 2 does not begin copying at OH in the NCR, but rather five eighths of the circle away, suggests the possibility that Copy 2 may not shorten either of its psephomere strands. Stuttering is seen only at the end of the synthesis of a strand, when a strand's two ends need to be joined together, and, for Copy 2, the psephomere is in the middle of the synthesis, not at the end (Figure 5).
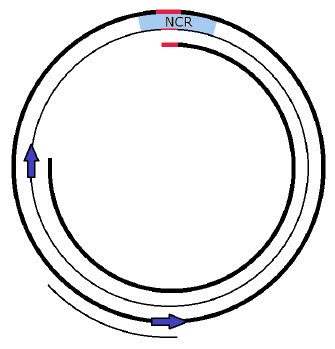 |
If this is correct, then the template L strand emerges as the "queen bee" strand. It produces "brood" strands, all of the same generation, one generation older than the queen.
To see this, focus on Copy 1 (e.g., the inner two tracks in Figure 5). On completion, Copy 1 contains a new H strand, but the queen (the template L strand) has survived and is now resident in the new Copy 1. This passage forward of the queen in the new Copy 1 may repeat itself over and over, for however many replications may be needed. This means that the template L strand is THE template, which gets used over and over. It makes a Copy 1 H strand which is always just 1 generation older than itself each replication, but then this H strand gets cast off as Copy 2 in the next replication. Thus, the Copy 1 L strand is the queen, which is capable of casting innumerable Copy 2 brood, all of the same generation number.
Because Copy 2 replicates its template H strand without shortening either strand of its psephomere, the new Copy 2 L strand psephomere winds up recording that it is one generation older than the queen. One may think of Copy 2 L strands as princesses—daughters of their queen mother.
Unfortunately, queens are not immortal. The template L strand is as much subject to free radical damage as any other L strand. When the queen is killed by free radical damage, she must be replaced. A new queen must be chosen from her mitochondrion hive. The logic of generation counting to this point implies that the mitochondrial hive will be inhabited only by sisters to the queen or by princesses. If a sister exists, then she needs to be chosen as the next queen since she will be one generation closer to the original blueprint than a princess. The eventual loss of all sisters will cause a princess to become queen. This increments the generation number of the queen and corresponds to the bursting of one balloon.
To choose the next queen, it is necessary to read and compare the psephomere L strands of all existing mtDNA copies in the mitochondrion. How is this to be accomplished? I suggest it may be accomplished individually and simultaneously for each copy by the normal mtDNA copying apparatus, POLγ. This postulate immediately explains why the loop is a three-stranded D-loop. To read the psephomere L strand, POLγ must scan it. So it scans the NCR L strand, and simultaneously synthesizes the complementary H strand, up to TAS. This creats a short, three-stranded region continuous with the initial loop—creating the D-loop. Once the psephomere L strands have been read, all POLγs of all mtDNA copies must wait until it is decided who is to be queen. This explains why TAS exists. TAS opens only to the new queen.
This new algorithm is incomplete, and it seems unlikely even that every detail of it which has been given is correct. For example, it seems at least possible, if not probable, that queen sisters cast brood simultaneously rather than sequentially. But the form of the overall process seems sufficiently clear and robust to render the vestiges discovered real rather than imaginary. Thus, the map of diseased mtDNA and its accompanying algorithm for replication of diseased mtDNA do indeed appear to exhibit psephomere vestiges. The curious idling delete cycles just when and where psephomere shortening might be anticipated, the unavoidable need of a loop to take up the slack once the Copy 1 psephomere H strand has been shortened, the need to scan the psephomere to read the generation number thus giving rise to a three-stranded D-loop region at the start of Copy 1, and the existence and placement of TAS sum to support the theory of Aging 2.
A mechanism of mtDNA failure giving rise to Aging 2 disease has now been explicated. This completes the two-phase theory of human aging, furnishing a complete explanation of what modern human aging is and how it comes about. Aging 0 is a nutritional deficiency disease of vitamin MePA. Aging 1 is a nutritional deficiency disease of vitamin MePiA. The cures of these two aging diseases are known and available. Aging 2 has now been explained as a malfunction of the copy error suppression mechanism for mtDNA, caused by psephomere exhaustion due to excessive copying of mtDNA, due, for example, to high concentrations of free radicals requiring frequent replacement of damaged mtDNA copies. Malfunction of the copy error suppression mechanism results in accumulating mtDNA point mutations, which results in declining OXPHOS ability and a host of other potential dysfunctions, the collective sum of which constitutes clinical Aging 2 disease, including, for example, heart attack and cancer. ◇
The Biblical Chronologist is written and edited by Gerald E. Aardsma, a Ph.D. scientist (nuclear physics) with special background in radioisotopic dating methods such as radiocarbon. The Biblical Chronologist has a fourfold purpose: to encourage, enrich, and strengthen the faith of conservative Christians through instruction in biblical chronology and its many implications, to foster informed, up-to-date, scholarly research in this vital field, to communicate current developments and discoveries stemming from biblical chronology in an easily understood manner, and to advance the growth of knowledge via a proper integration of ancient biblical and modern scientific data and ideas. The Biblical Chronologist (ISSN 1081-762X) is published by: Aardsma Research & Publishing Copyright © 2020 by Aardsma Research & Publishing.
|
^ Gerald E. Aardsma, "Actuarial Data Show That Vitamin MePA Deficiency Disease Is Needlessly Killing a Quarter Million Americans Annually," The Biblical Chronologist 10.15 (October 28, 2020): 1–16. www.BiblicalChronologist.org.
^ Gerald E. Aardsma, "Human Aging is a Two-Phase Disease," The Biblical Chronologist 10.8 (May 13, 2020): 7. www.BiblicalChronologist.org.
^ draardsmasvitamins.com/get-the-vitamins/
^ Gerald E. Aardsma, "Human Aging is a Two-Phase Disease," The Biblical Chronologist 10.8 (May 13, 2020): 6–8. www.BiblicalChronologist.org.
^ Gerald E. Aardsma, "Human Aging is a Two-Phase Disease," The Biblical Chronologist 10.8 (May 13, 2020): 7. www.BiblicalChronologist.org.
^ Gerald E. Aardsma, Addendum to Aging: Cause and Cure (Loda, IL: Aardsma Research and Publishing, July 26, 2019), 10–12. www.BiblicalChronologist.org.
^ Gerald E. Aardsma, "Human Aging is a Two-Phase Disease," The Biblical Chronologist 10.8 (May 13, 2020): 8. www.BiblicalChronologist.org.
^ www.yourgenome.org/facts/what-is-a-telomere (accessed 2020/11/07).
^ Gerald E. Aardsma, "Human Aging is a Two-Phase Disease," The Biblical Chronologist 10.8 (May 13, 2020): 8. www.BiblicalChronologist.org.
^ This observation leads immediately to the postulate that the phylogenetic development of a self-replicating, mutating machine will be grainy rather than smooth. While mutation of the code is likely to be a gradual, smooth process, the effect of code mutations on the coded idea is likely to be grainy and has potential even to be discontinuous.
^ Maria Falkenberg, "Mitochondrial DNA replication in mammalian cells: overview of the pathway" Essays in biochemistry 62.3 (July 20, 2018): 287–296. I have used this paper as my primer on the topic of replication of human mtDNA. It was written to "give a brief overview of DNA replication in mammalian mitochondria, describing our current understanding of this process and some unanswered questions remaining," a purpose to which it succeeds admirably, in my opinion.
^ Personal communication October 25, 2020.
^ For the current map of human mtDNA, see: Maria Falkenberg, "Mitochondrial DNA replication in mammalian cells: overview of the pathway" Essays in biochemistry 62.3 (July 20, 2018): 287–296, Figure 1.
^ Gerald E. Aardsma, "Human Aging is a Two-Phase Disease," The Biblical Chronologist 10.8 (May 13, 2020): 7. www.BiblicalChronologist.org.
^ Gerald E. Aardsma, "Actuarial Data Show That Vitamin MePA Deficiency Disease Is Needlessly Killing a Quarter Million Americans Annually," The Biblical Chronologist 10.15 (October 28, 2020): 10. www.BiblicalChronologist.org.
^ medlineplus.gov/genetics/chromosome/mitochondrial-dna/ (accessed 2020/11/03).